Genomics and Applied Biology 2018, Vol.9, No.10, 62-71
67
classifies sequences based on their characteristics. This software calculates the replacement frequency of codons
adjacent to the transcripts of coding protein and non-coding protein, which is later used to construct score matrix.
The most similar coding sequence (CDS) are selected, and SVM classifier is constructed by combining single
nucleotide frequency. This software works for incomplete sequences, so it is more suitable for partial EST
sequences or transcripts spliced from scratch.
At present, there are many other prediction software in addition to the two kinds of prediction software described
above. The core ideas of these software are the same, but the specific implementation methods are different.
According to the different cases of specific sequence, the predicted results of these software have their own
advantages and disadvantages, and the intersection of the predicted results of several software can be used as a
reliable result.
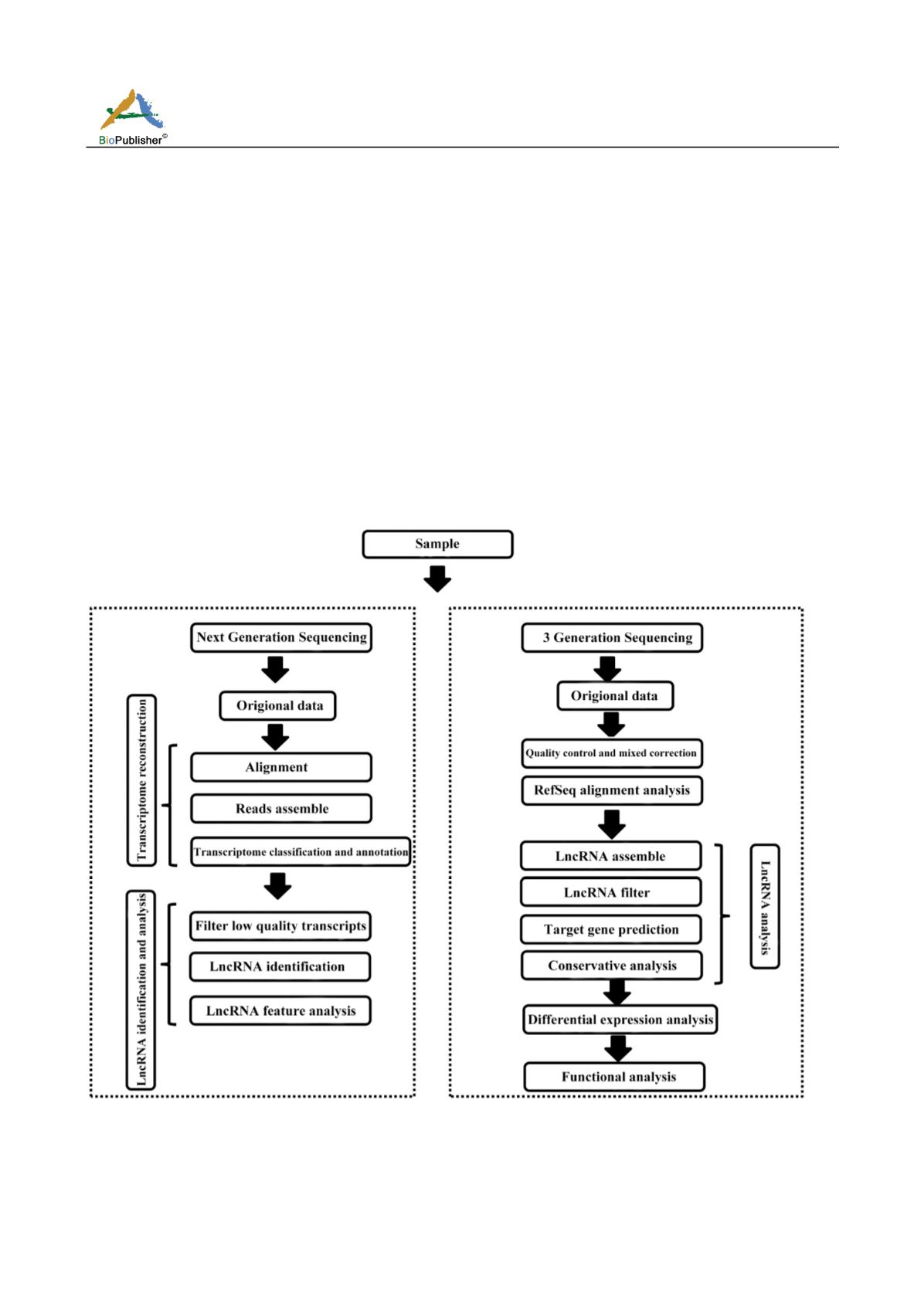
6 Bioinformatical Identification Procedure of LncRNA
Now the analysis and identification process of lncRNA can be roughly divided into two categories according to
their data sources (Figure 4). The data generated by Illumina sequencing technology can be identified and
analyzed in one category. Another category is to identify and analyze the data obtained by single-molecule
sequencing with third-generation sequencing technology.
Figure 4 Flow diagram of lncRNA identification by next
generation sequencing data and three generation sequencing data
Illumina sequencing platform is widely used. The data generated by Illumina sequencing platform need to be
analyzed and converted into original sequence data (Raw Data). The next thing is to filter and clean the data,
because the raw offline data generally contains some joint contamination, and may also contain some low-quality
reading lengths. The general requirements of filtration operations are to remove reads with sequencing connectors


