巩鹏涛等:蛋白质基因组学研究中的质谱仪与生物信息学方法
Copyright © 2015 BioPublisher Jisuan Fenzi Shengwuxue | Vol.4 | No.1 | 1–12
Galaxy-P
分析的灵活性的体现之一就是数据库
构建的灵活性,使得
Galaxy-P
可以针对不同的样品
进行不同的数据库改变。这种灵活性带来便利和针
对性在研究者使用该分析流程对人类唾液的蛋白
质基因组学的演示分析中得到体现。研究人员发现
在
PSMs
搜索分析中使用人类蛋白质数据库加入人
类口腔微生物的蛋白质数据库组成综合数据库,比
单独使用人类蛋白质数据库要多发现两倍的新序
列变异体。导致这一问题出现的原因就是:微生物
序列的缺失会导致串联质谱得到的来自非宿主的
肽被迫和宿主的蛋白质相匹配,增加了假阳性并迫
使
PSMs
为得到可接受的
FDR
必须有一个更高的得
分值,因此就降低了新肽序列可信匹配的数量。这
一发现说明在进行蛋白质基因组学的研究中,在样
品包含非宿主的蛋白质情况下进行的蛋白质基因
组学分析,如果仅仅使用宿主的蛋白质序列进行数
据库搜索,其他结果会受到很大影响。
Galaxy-P
灵活性的另外一个表现就是搜索时可
以使用不同的质谱数据库搜索引擎,这样可以相互
验证并弥补不足。在演示分析研究中
ProteinPilot
和
X!Tandem
的使用就充分证明了这一点,另外
SearchGUI (Vaudel et al., 2011)
未来整合入
Galaxy-P
的蛋白质基因组学分析流程就更值得期待。序列
数据库的搜索中,
“
明尼苏达两步法
”
的使用也可
以解决蛋白质基因组分析中大蛋白质数据库所固
有的挑战
(Jagtap et al., 2013)
。在两步法中,第一
步的数据库搜索使用比较宽松的严谨度来鉴定那
些最可能在样品中存在的蛋白质,组成一个比较
小的蛋白质库;在第二步中对第一步产生的修正
的更小蛋白质数据库和与其相匹配的质谱被施加
了高度严密的条件进行二次分析,在可接受的
FDR
水平产生
PSMs
。
蛋白质基因组学分析往往依靠单个的
PSMs
来
确定潜在的新蛋白序列,考虑到单个
PSMs
带来的
潜在的假阳性,
Galaxy-P
的蛋白质基因组学分析流
程中提供了多中水平的质量控制和过滤。除了在数
据库搜索模块使用多个数据搜索引擎来改善结果
的可信度外,在关键的第三模块中
BLASTP
方法的
使用和自行开发的
PSME (peptide spectrum match
evaluation)
工具是
PSMs
质量控制的重要一环。
BLASTP
中和
NCBI
数据库的比对可以进一步过滤
掉和已知序列匹配的
PSMs
。
PSME
不仅提供了一
种可视化串联质谱图谱及其对应假定序列匹配的
工具,还可以用户自行设定多种
PSM
质量相关的
参数来对质谱进行过滤。在
Galaxy-P
演示数据的
分析中
BLASTP
的分析将
9333
个
PSMs
减少点
1630
个,
PSME
高严谨度
PSM
质量标准的限定更
是将新肽序列匹配的数量减少到
55
个。
Galaxy-P
的蛋白质基因组学分析流程的最后一
步是通过自开发的“
Peptides to GFF
”工具来将肽
段的氨基酸序列转换为
IGV (integrated genome
viewer)
兼容的格式,从而实现在基因组中的可视化
和阐释。新肽片段在基因组上的可视化有利于进一
步的对这些肽段所对应假定新蛋白质可能性的评
估分析。
虽然,基于
Galaxy-P
的蛋白质基因组学分析流
程尽管包含大约
140
多个步骤,但整个流程在参数
优化设定的情况下,只需要一次单击就可以完成整
个流程,最要的是每个模块中的分流程都可以按需
求单独自我运行。这些整个流程可以通过一个网络
连接或一个保存的
Galaxy
流程文档而与其他人分
享,和流程文档一样
Galaxy-P
的历史文档也可以被
分享,历史文档中包含了重现整个分析流程所需要
的所有的软件和各种输入与输出数据。
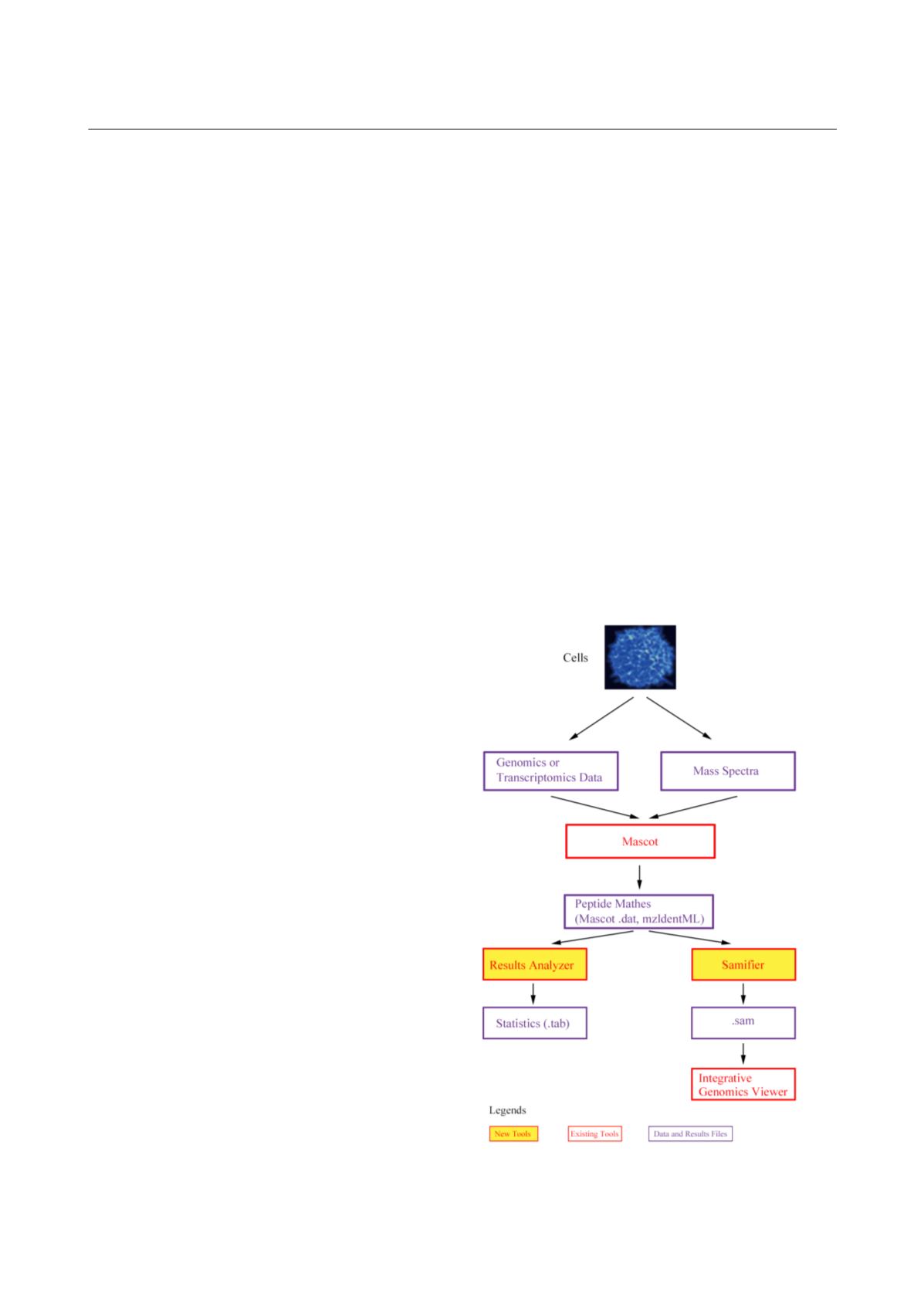
2.10 Proteomic-Genomic Nexus
Proteomic-Genomic Nexus
是基于
Java
语言软
件包,其设计目的是将下一代测序产生的基因组和
转录组数据和蛋白质质谱产生的蛋白质组数据进
行整合,该软件包的具体分析流程可见图
4
。
图
4 The Proteomic-Genomic Nexus
分析流程图示
Figure 4 The Proteomic-Genomic Nexus analysis workflow


