基本HTML版本



Genomics and Applied Biology 2014, Vol. 5, No. 5, 1-6
http://gab.biopublisher.ca
5
A total of 629 unigenes were annotated with 89
pathways in the KEGG database (http://www.genome.
jp/kegg/pathway.html). Many transcripts include
various pathways like metabolic pathways,
plant-pathogen interaction pathways, fatty acid
metabolism pathway and fatty acid biosynthesis.
2.4 SSR mining
Microsatellite markers (SSR markers) are some of the
most successful molecular markers in the construction
of a
Phaseolus vulgaris
L. genetic map and in
diversity analysis (Zhang et al). For identification of
SSRs, all transcripts were searched with perl script
MISA. We identified a total of 1405 SSRs in 1304
transcripts (Table 5). The mono-nucleotide SSRs
represented the largest fraction of SSRs identified
followed by tri-nucleotide and di-nucleotide SSRs.
Although only a small fraction of tetra-, penta- and
hexa-nucleotide SSRs were identified in transcripts,
the number is quite significant.
Table 5 Statistics of SSRs identified in transcripts
SSR Mining:
Total number of sequences examined:
6999
Total size of examined sequences (bp):
2110290
Total number of identified SSRs:
1405
Number of SSR containing sequences:
1304
Number of sequences containing more than one SSR: 86
Number of SSRs present in compound formation: 64
Distribution to different repeat type classes:
Mono-nucleotide
1218
Di-nucleotide
87
Tri-nucleotide
90
Tetra-nucleotide
7
Penta-nucleotide
2
Hexa-nucleotide
1
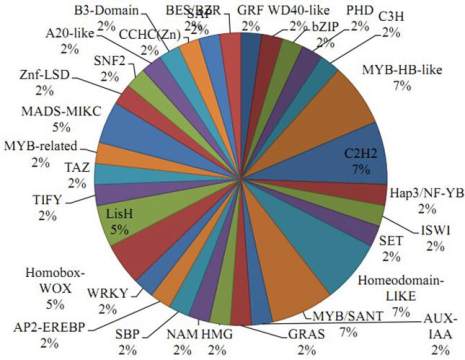
2.5 Plant Transcription Factor
Further, transcription factor encoding transcripts were
identified by sequence comparison to known
transcription factor gene families. Result shows that
transcription factor genes distributed with at least 32
families were identified (Figure 5). The overall
distribution of transcription factor encoding transcripts
among the various known protein families is very
similar with that of other legumes as predicted earlier
(Libault et al., 2009).
Figure 5 Plant Transcription Factor Result
3 Conclusion
This study is focus on
Phaseolus vulgaris
L. species
(SRR1283084) from NCBI database for de novo
Transcriptome analysis by RNA-seq using next-generation
Illumina sequencing. The transcriptome sequencing
enables various functional genomics studies for an
organism. Although several high throughput
technologies have been developed for rapid sequencing
and characterization of transcriptomes, expressed
sequence data are still not available for many
organisms, including many crop plants. In this study,
we performed de novo functional annotation of the
Phaseolus vulgaris
L. transcriptome without considering
any reference species with significant non-redundant
set of 6999 transcripts. The detailed analyses of the
data set has provided several important features of
Phaseolus vulgaris
L. transcriptome such as GC
content, conserved genes across legumes and other
plant species, assignment of functional categories by
GO terms and identification of SSRs by MISA tool. It
is noted that this study of
Phaseolus vulgaris
L. will be
useful for further functional genomics studies as it
includes useful information of each transcript.
Acknowledgement
We are heartily thankful to Prof. (Dr.) P.V. Virparia, Director,
GDCST, Sardar Patel University, Vallabh Vidyanagar, for
providing us facilities for the research work.
References
Collins J. L., Biggs J. P., Voelckel C., and Joly S., 2008, An approach to
transcriptome analysis of non-model organisms using short-read