基本HTML版本



Computational Molecular Biology 2014, Vol. 4, No. 10, 1-17
http://cmb.biopublisher.ca
3
1.2 Peak finding
The concept of co-localization is based on identified
peak in this study. The ChIP-seq peak finding
procedure is illustrated as below. A negative binomial
model for each modification profile was trained to
provide FDR control, for the negative binomial model
provides a much better fit to the ChIP-seq data than
does the Poisson model. FDRs were estimated by
modeling the read count in windows using negative
binomial distribution. Each chromosome was scanned
with the window size of 100bp with window moving
consecutively per 25bp. Under the negative binomial
model, windows with read counts greater than a user
chosen cutoff for
bona fide
binding regions were
identified by controlling FDR < 0.05.
1.3 Co-localization peak identification
To classify genome-wide peaks into different
co-localized groups: me1me2, me1me2me3, me1me3,
me2me3 and controls (single-localized peaks),
genomic intervals were compared exhaustively. Ten
Overlap rate (OR) cutoffs were considered in parallel.
OR = 1.0 is the most stringent co-localized peak cutoff,
likewise, OR = 0.1 generates the loosest. Most
analysis in this study took OR = 0.5 as a basis if no
explicit statement was declared.
1.4 Gene overlapping analysis
To assess the functional genomic attributes for peaks,
we associated the co-localized and single-localized
peaks with TPRs defined by upstream 1k and
downstream 2k around TSSs of any annotated genes.
The boundaries for TPRs were suggested by the study
of Barski et al.
1.5 Gene Ontology enrichment analysis
RefSeq mRNA IDs of co-localized peaks overlapping
with annotated genes were submitted to the DAVID
system
. Only GO terms with
reported
p
-values smaller than 10E-3 and met by
Bonferroni multiple testing correction cutoffs were
extracted.
1.6 Motif analysis
It was interesting to search for enriched sequence
patterns for four classes of co-localized peaks
overlapping with genes. We used Gibbs Motif
Sampler with 3000 iterations powered by cisGenome
suite to perform the analysis
. For the
generated motifs, only one key motif was considered
as the enriched one by performing motif enrichment.
To make a fair control, the matched genomic control
sequences simulated from corresponding co-localized
peak sequences were used. The software configuration
was set according to online tutorial.
2 Results
2.1 Genomic element distribution for different
localization types
A total of 82,283 peaks were identified by peak
detection. When considering the peak percentages in
TSS-proximal regions (TPRs, defined by upstream 1k
and downstream 2k around TSSs) and non-TPR
regions, we find co-localized peaks vary little in the
two regions (Supplementary Table 1). Generally, Me1
localizes less in TPRs, while me2 and me3 localize
more in TPRs compared with non-TPRs. The number
of me2me3 co-localization is the most relative to other
co-localization types (6% overall).
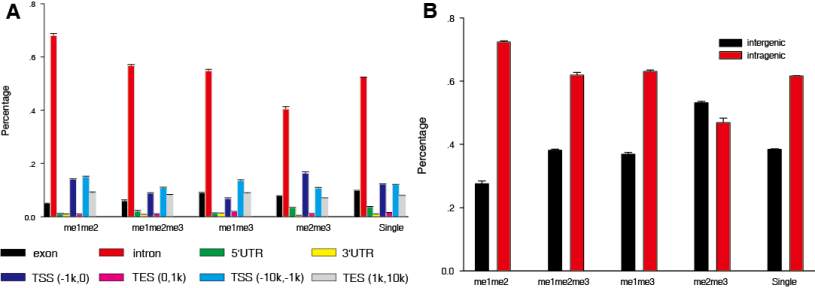
Figure 1 Genomic distribution of co-localized peaks. (A) Distribution of co-localized peaks within gene context. RefSeq genes were used
as the gene annotation reference, the genomic annotation is from UCSC Table Browser. (B) Distribution of co-localized peaks and
single-localized peaks across genome, where transcriptional Start Sites (TSSs) and Transcriptional End Sites (TESs) are gene boundaries