基本HTML版本



International Journal of Clinical Case Reports 2014, Vol. 4, No. 4, 1-3
http://ijccr.biopublisher.ca
2
CT scan found a slight triventricular passive
hydrocephalus.
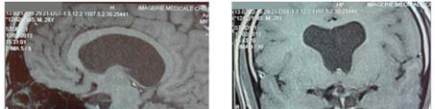
MRI showed an aspect of
holoprosencephaly with dilatation of the lateral
ventricles, absence of the septum pellucidum and a
septo-optic dysplasia (Figure 1).
Figure 1 MRI showing an holoprosencephaly aspect
The patient received growth hormone treatment
reaching a height of 169 cm after 5 years of treatment.
Hypogonadism was treated with androgens with a
good development of secondary sexual characters.
3 Discussion
There are 4 forms of HPE with variable severity
according to the degree of non separation of the
cerebral hemispheres: alobar (complete fusion of CH),
semilobar (fusion of frontal lobes), lobar (fusion of the
basal part of the frontal lobes), Interhemispheric or
syntélencéphalie (fusion of the posterior part of the
frontal lobes and the parietal lobes) (Hahn and Barnes,
2010).
The Septo-optic dysplasia as described in our patient
is now recognized as a form of lobar HPE, with
aplasia of the septum pellicidum, moderate dilatation
of the lateral ventricles with rectangular appearance of
frontal horns, a hail aspect of the optic chiasma with
presence of a ventral inter-hemispheric fissure of the
frontal lobes (unlike the classic lobar form).
HPE is associated with craniofacial abnormalities
related to the degree of non separation of CH
(DeMyer et al., 1964). Signs that may be encountered
ranging from cyclopia to normal facies or almost (Xin
and Guillermo, 2009), as in our patient who just has a
wide flat nose.
HPE can be caused by (Muenke et al ., 2001):
-Structural or numerical chromosomal abnormality
-Exposure to environmental factors and teratogens in
utero as maternal diabetes, alcoholism, smoking,
drugs and maternal infections
-Be part of a syndrome (Smith-Lemli-Opitz syndrome,
Pallister Hall, Rubinstein-Taybi, Meckel etc.)
In our patient we only noted maternal diabetes, that
occurred few years after birth, this does not exclude
the possibility of a gestational diabetes participating in
the genesis of HPE.
Finally there is the isolated forms or
non-chromosomal, non syndromic, due to a mutation
in one of the 12 known genes. Genetic forms are
inherited as an autosomal dominant forms with highly
incomplete penetrance and variable expressivity
(Benjamin, 2010).
The clinical picture varies according to CNS
structures involved. Functions controlled by the
hypothalamus such as thermal regulation, thirst,
appetite, sleep and blood pressure may be affected, all
functions seem preserved in our patient, but close
attention should be paid during the follow-up to detect
any disturbance. The pituitary gland is not spared
(Hahn et al., 2005), abnormalities can be structural or
functional as in our case. Diabetes insipidus is more
frequent than anterior pituitary deficits in HPE due to
the hypothalamic origin of the cores regulating hydric
balance.
Neurological signs include motor dysfunction
(hypotonia, dystonia, spasticity), epilepsy (50% of
patients) and hydrocephalus. Delayed psychomotor
development and mental retardation are also observed
in our patient and classically described in HPE.
The prognosis is closely related to the degree of brain
malformation:
The alobar and semi lobar forms have a dire prognosis
(death within few weeks or few months of life). Other
forms like our patient, have a better prognosis and can
expect an almost normal life provided an adequate
monitoring and management of their medical
problems involving a multidisciplinary team with an
endocrinologist, a neurologist, an ophthalmologist a
and a neurosurgeon (Barr and Cohen, 1999).