基本HTML版本


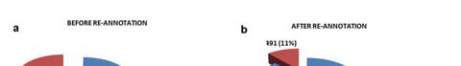
I
n silico Proteomic Functional Re-annotation
Escherichia coli
K-12 using Dynamic Biological Data Fusion Strategy
36
powerful re-annotation. In silico re-annotation permits
uniform quality control, systemic updates, easy data
parsing and more comprehensive comparative analysis,
providing a valuable resource to the whole research
community (Rajadurai et al., 2011). Though, several
genome and proteome database are publicly available
to facilitate life science research, there are a number of
pitfalls associated with each database. The most
common problems in these biological sequence
databases are lack of reliability, redundancy, erroneous
annotation etc. Recently, the misannotation levels in
the four popularly used public protein sequence
databases, UniProtKB/Swiss-Prot, GenBank NR,
UniProtKB/TrEMBL,
and KEGG have been
investigated and identified the misannotated enzyme
superfamilies that remains a larger problem during
annotation process (Schnoes et al. 2009).
To alleviate the problem of erroneous annotation and
redundancy, re-annotation of genes and proteins using
a set of common, controlled context to describe a gene
or protein function is necessary. Thus, deciphering the
precise functions encoded by all gene products of this
genome remains a great challenge in this genomic era
(Bock and Gough, 2004; Altschul et al., 1990). Hence,
to overcome such challenges, the re-annotation of the
E. coli
K-12 proteome has been carried out using a
strategy known as “dynamic biological data fusion” in
which the biological data from various available
databases are integrated into a unique information
source. Further, confidence level has been carried out
to assign functions of unknown proteins and thereby
facilitating more accurate functional information to
the research society.
1 Results
The original sequence annotations of
E. coli
K-12
strain downloaded from EcoCyc database and it was
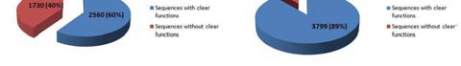
identified to possess 4,290 protein sequences. Of these
sequences, 2,560 sequences had clear annotations and
the remaining 1,730 sequences were found to be
uncharacterized with hypothetical,
unknown,
predicted, conserved and putative functions (Table 1,
Figure 1a). Following the in silico functional
re-annotation, several categories of changes were
made in the previously annotated E. coli genome that
includes i) Assigning functions to uncharacterized
proteins ii) Transfer of functions (revision of already
annotated function) and iii) Updating the functions.
Figure 1 EcoCyc Genome Data
Note: a. A pie chart describing the percentage of known and
unknown sequences in the original data downloaded from
EcoCyc. b. Genome data after re-annotation. A pie chart
representing the percentage of known and unknown sequences
after re-annotation (Rec-DB data)
Table 1 Genome features of
E. coli
K-12
Sequence Category
Number of Sequences
Percentage (%)
Total No. of protein Sequences
4290
100
i. Sequences with unknown functions:
Predicted
1033
24.08
Conserved
401
9.35
Putative
212
4.94
Conserved + Hypothetical
18
0.42
Hypothetical
59
1.38
Conserved + Predicted
2
0.05
Conserved + Putative
1
0.02
Hypothetical + Predicted
2
0.05
Putative + Predicted
2
0.05
Total Sequences with unknown functions
1730
40
ii. Sequences with clear functions
2560
60
Computational
Molecular Biology